
薬学系の人でも知らない気がするので。
この記事には広告が含まれています。
PMDAで新薬審査してました
薬学系の博士課程を修了後、独立行政法人医薬品医療機器総合機構(PMDA)に就職し、新薬審査に携わっていました。
過去形なのは先日退職したからで、現在はヘルステック業界に転身し、ITツールを使って医療アクセスが十分でない地域の人々に専門医の診療を届けられるような仕組み作りをしています。
現在進行形の仕事だったのでこれまでブログでは触れてきませんでしたが、退職して外部の人間になったので、PMDAの新薬審査部で行っている仕事についてご紹介したいと思います。
なお、この記事は2019年10月に僕が出身大学で講演したときのスライドを元に構成しています。
もくじ
PMDAとは
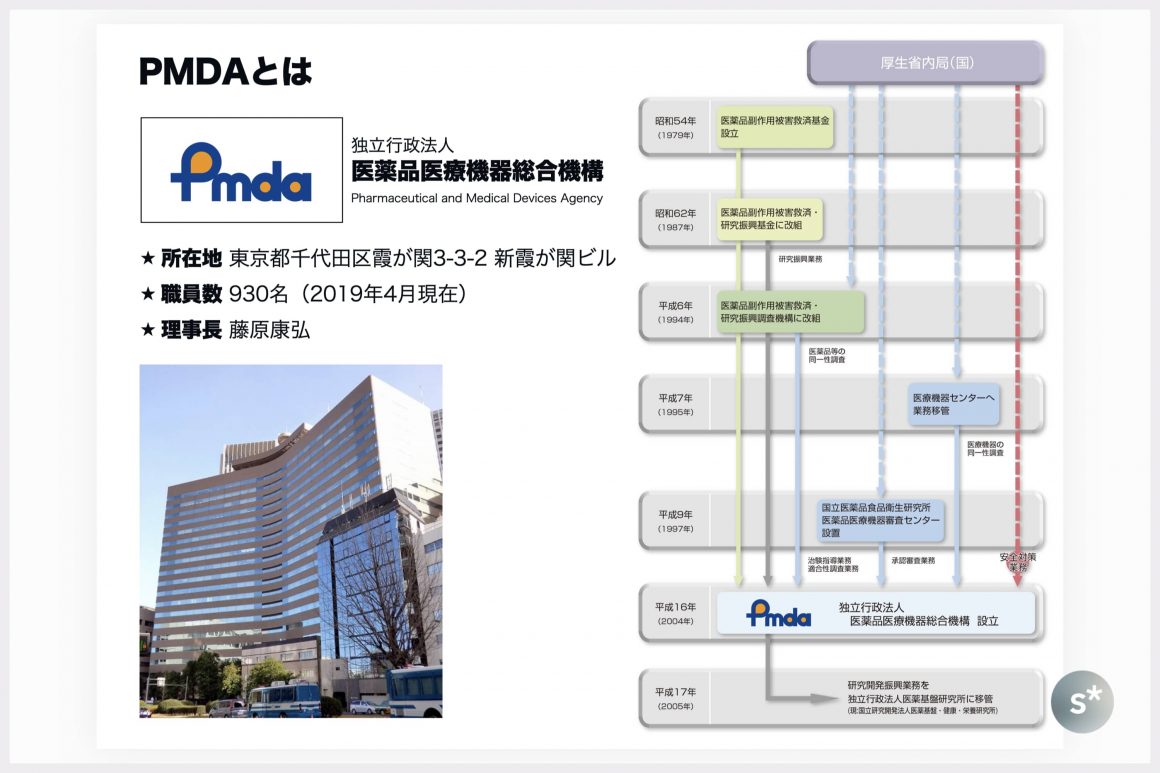
薬学に携わっている人ならご存知だと思いますが、念のためPMDAについて説明しておきます。
正式名称は「独立行政法人医薬品医療機器総合機構」で、英語名の「Pharmaceutical and Medical Devices Agency」の頭文字を取ってPMDAと略されます。厚生労働省管轄の独立行政法人で、複数の組織が統合されて2004年に発足しました。
アメリカでのFDA、ヨーロッパでのEMAというように、世界各地に同様の規制当局がありますが、PMDAは日本における唯一の薬事に関する規制当局です。
場所は東京都千代田区霞が関で、総理大臣官邸の目と鼻の先にあります。職員数は、2021年現在は1,000人を少し超えた程度と思います。
PMDAの業務
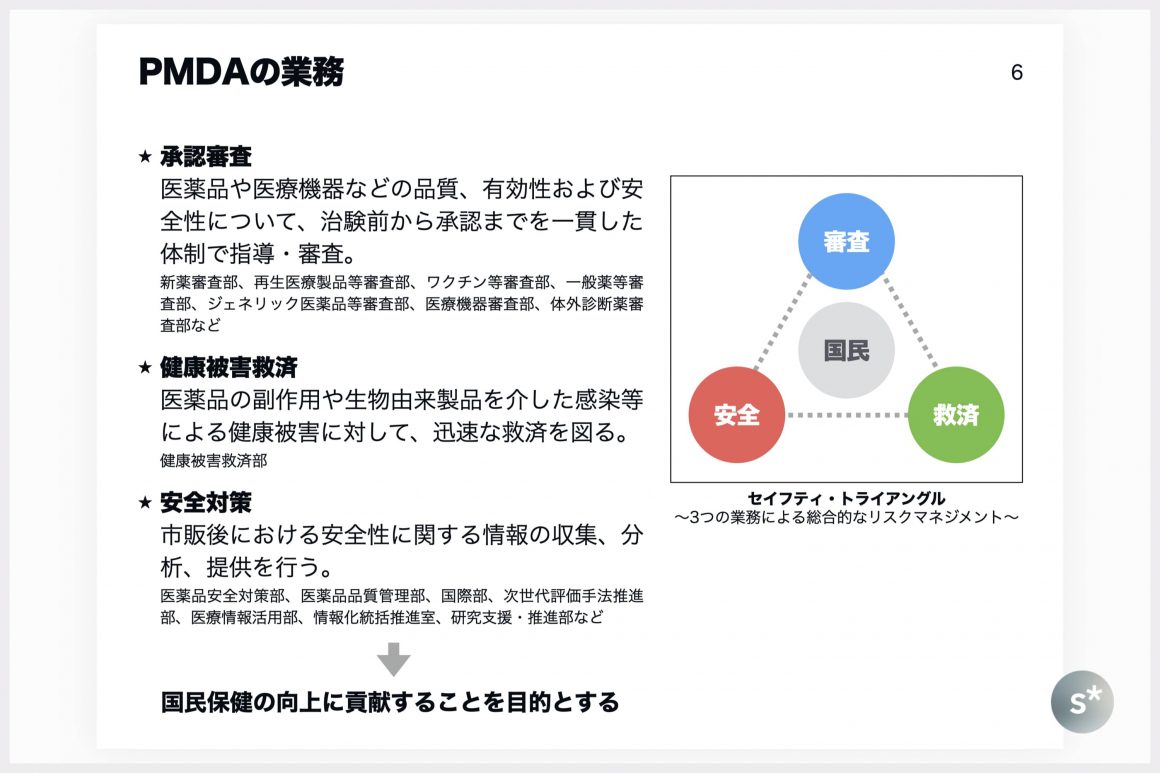
PMDAの仕事は「審査」「安全」「救済」の3つに分かれており、セイフティ・トライアングルと呼ばれています。これら3つの業務を進めることで、国民保健の向上に貢献することを目的として、日々活動しています。
「審査」とは承認審査のことで、医薬品や医療機器は厚生労働大臣から承認されないと日本国内で販売することはできません(医療機器はクラス分けされており、クラスによっては都道府県の管轄)。そのための審査をPMDAで行っているのです。
「救済」とは、医薬品の副作用や生物由来製品を介した感染による健康被害に対して、患者さんを補償します。補償金は製薬企業などからの拠出金でまかなわれています。医薬品の審査を行う規制当局が救済まで行うのは世界的にも珍しいそうです。
「安全対策」とは、医薬品が承認され、販売開始後において情報の収集、分析、提供を行います。治験では明らかにならなかった副作用やその他の有害事象が現れていないか調査し、販売開始後の医薬品の安全性を司ります。
以上のような3つの業務をもって、「国民保健の向上に貢献することを目的とする」とされています。根拠法は独立行政法人医薬品医療機器総合機構法です。
これらの業務の中から、僕が携わっていた新薬審査について説明します。
新薬審査業務

審査業務は、まず医薬品と医療機器に大別されます。医薬品の審査は、新薬審査第一部〜第五部、再生医療製品等審査部、ワクチン等審査部、ジェネリック医薬品等審査部、一般薬等審査部の各部署で担っています。
これらのうち、僕が携わっていた新薬審査においては、分子標的薬などを除く「化成品」の新薬を取り扱います。さらに、対象となる疾患領域ごとに第一部〜第五部まで分かれています。
また、新薬審査部といっても医薬品の承認審査だけを行っているわけではありません。後述しますが、「対面助言」とよばれる治験相談や、法律(薬機法)に則った治験届調査、学会参加なども行います。
新有効成分医薬品の審査期間
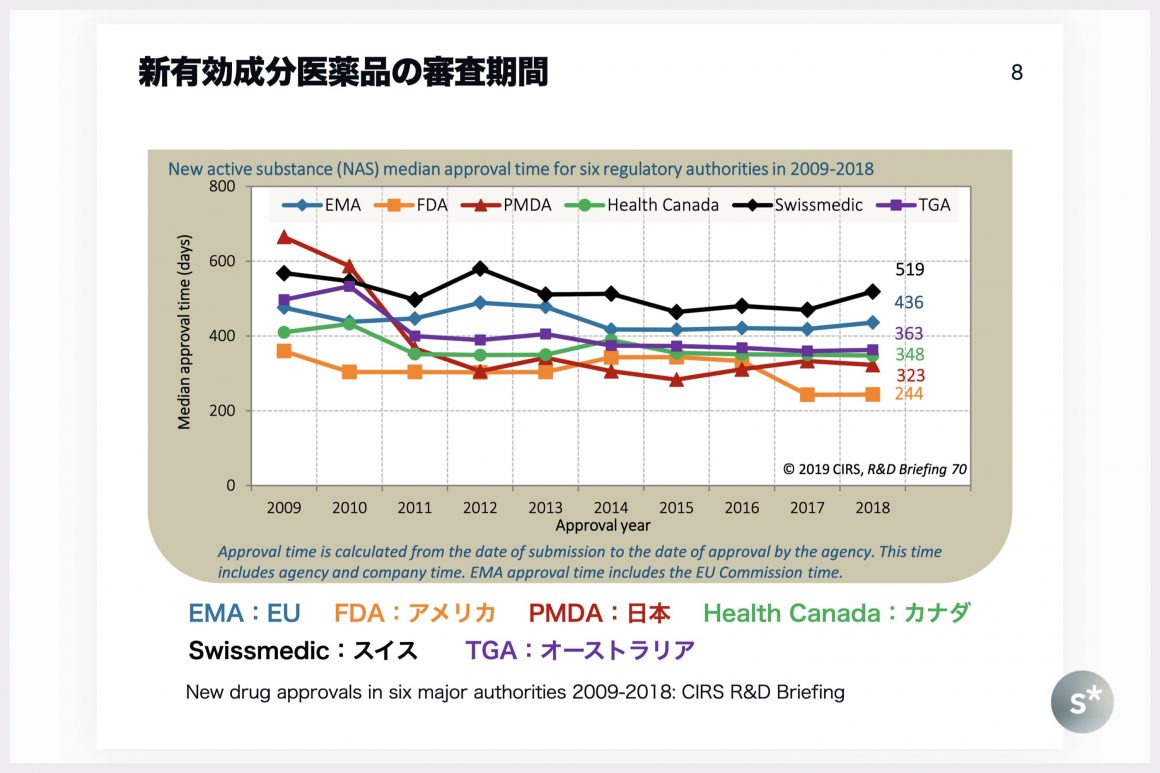
上の図で表しているのは、主要な規制当局における新有効成分医薬品の審査期間です。PMDAは赤で示しています。縦軸の値が小さいほど審査期間が短いということを表しており、2014〜2016年はPMDAが1位でしたが、その後FDA(アメリカ)が再度1位となっています。
PMDAにおける最近の審査品目はだいたい1年くらいでスケジュールを組んで回していますが、希少疾病用医薬品(オーファン薬)だともう少し早くて9か月でやっています。希少疾病用医薬品なので優先的に審査するという建前であるものの、実質的にはスケジュールを早回ししているだけなので、現場の負担はかなり大きいように思います。
さて、薬学を学んでいると「ドラッグラグ」という言葉を耳にしたことがあるかもしれません。このドラッグラグには「開発ラグ」と「審査ラグ」の2つがあると言われています。
- 開発ラグ —— 製薬企業が実施する治験の過程におけるラグ
- 審査ラグ —— PMDAの審査における海外規制当局とのラグ
上の図のように、PMDAの審査スピードは世界で1〜2位を争うくらいなので、審査ラグはほぼ解消されているような状況です。では、開発ラグの方はどうでしょうか?
臨床試験の流れ
開発ラグについて述べる前に、前提の共有を。
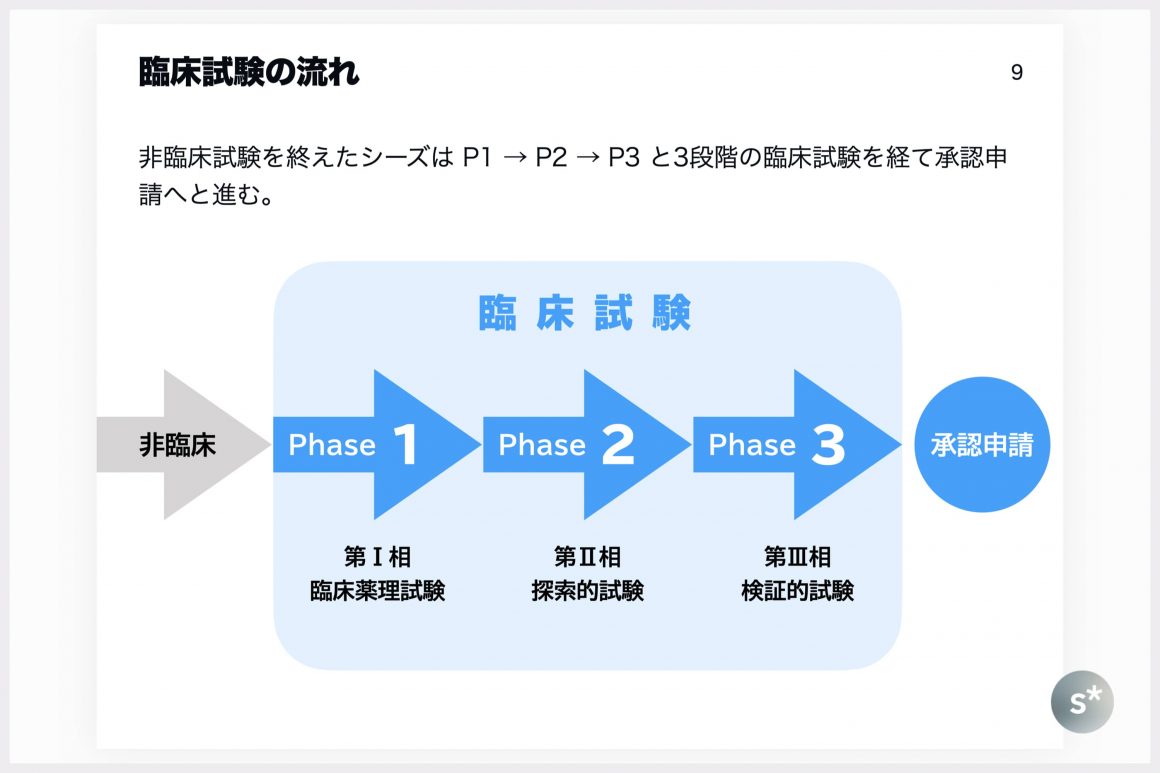
薬学を学んだ方ならご存じだと思いますが、非臨床試験を終えたシーズは臨床試験(治験)へと進みます。臨床試験はPhase 1〜3の3つの相に分かれており、それぞれの相で以下のような検証を行います。
- 第Ⅰ相試験 —— 薬物動態や安全性を確認する。
- 第Ⅱ相試験 —— 対象疾患の患者に投与して有効性・安全性を小規模で検討する。
- 第Ⅲ相試験 —— 第Ⅱ相試験よりも規模を拡大し、患者を対象に有効性・安全性を確認。また、長期的に投与されることが想定される薬剤の場合は、半年〜1年の長期投与時の安全性を確認します。
以前の新薬開発:国ごとの臨床試験
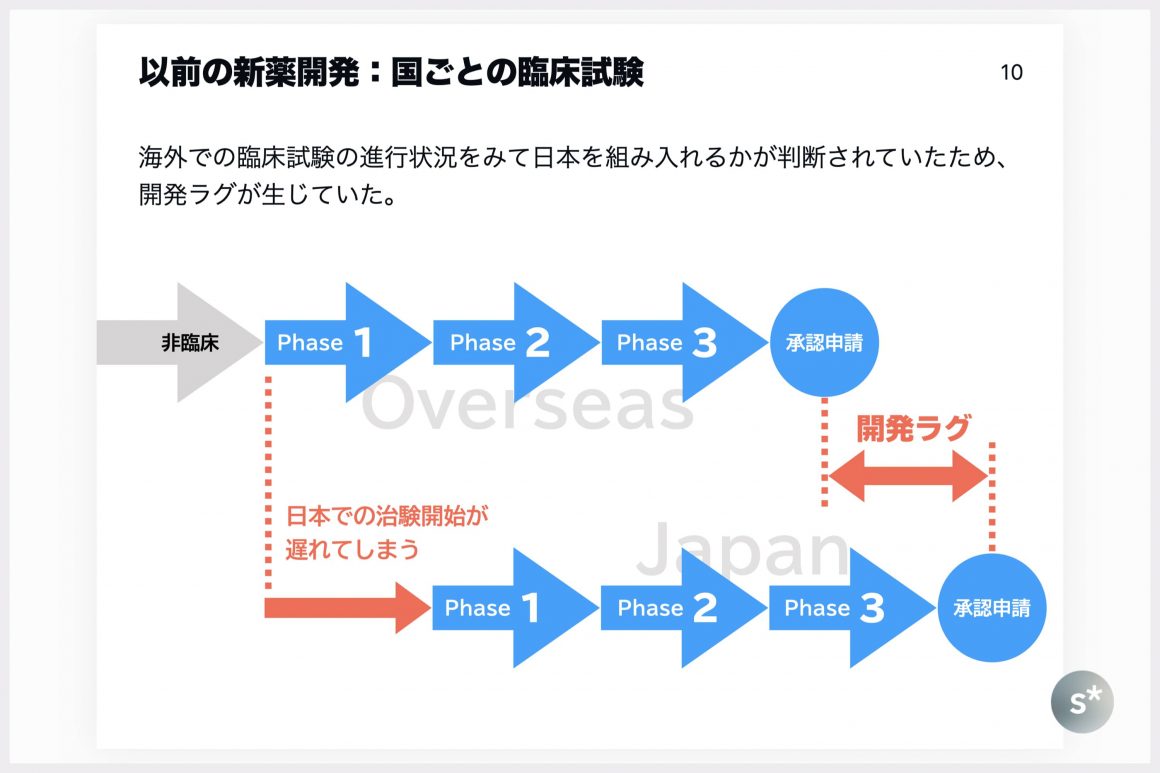
開発ラグが生じている原因として、「日本での臨床試験の開始が後回しにされてしまう」ということがあります。
臨床試験には多額の資金が必要なので、製薬企業としてはできる限り無駄な試験は実施したくないものです。欧米の製薬企業においては、まずは欧米で開発を進めておき、効果がありそうな場合に日本での開発を始めることがあります。
この場合、国ごとに臨床試験を実施すると「欧米での第Ⅰ相試験の開始から日本での第Ⅰ相試験の開始までのラグ」がそのまま「承認申請のラグ」になってしまいます。これを「開発ラグ」と呼びます。
最近のトレンド:国際共同治験
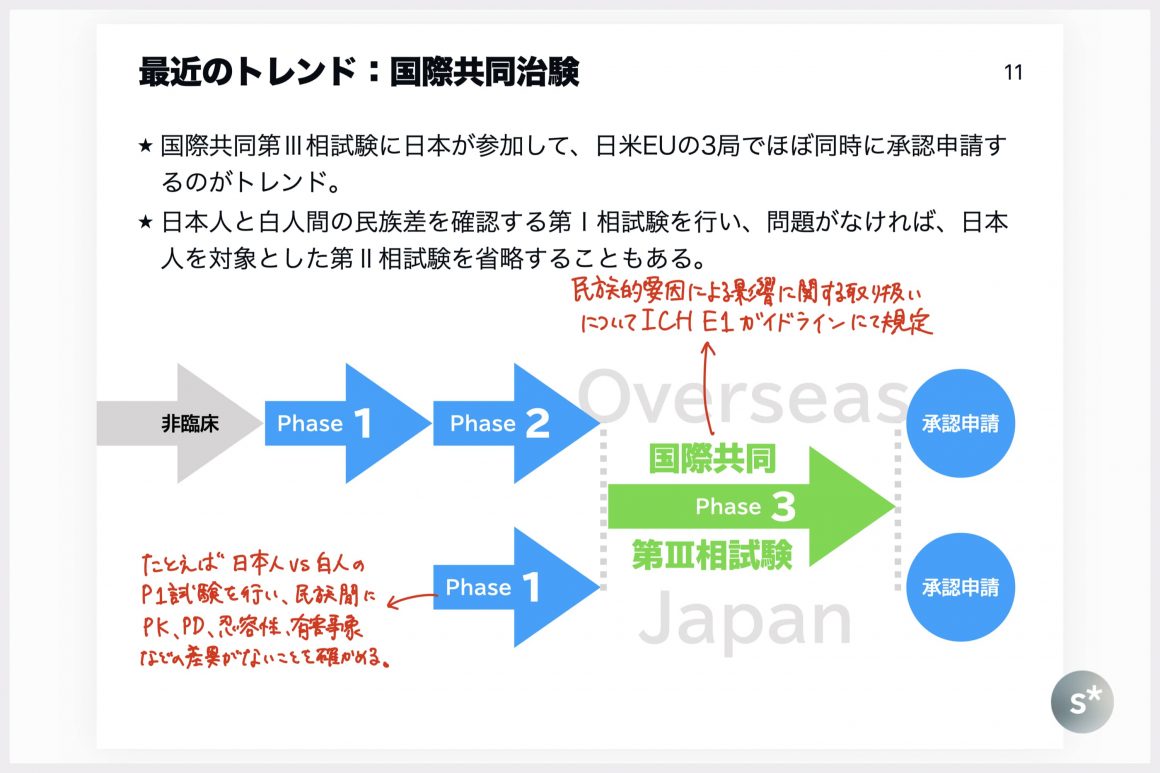
世界中の病気で苦しんでいる人々に早く薬を届けるという観点からは、開発ラグが生じている状況は好ましいものではありません。そのため、最近では第Ⅲ相試験を「国際共同治験」として実施することが多いです。これは、欧米人も日本人も一緒の試験の中に組み入れ、有効性・安全性を同時に見ていくという手法です。
かなり極端な例を上のスライドに記しました。欧米から開発を始め、日本は後回しになっている状況と仮定した場合、国ごとに開発すればラグが生じます。それを防ぐために、例えば、欧米の第Ⅰ相試験の結果と日本の第Ⅰ相試験の結果を比較して問題がなければ、日本人対象の第Ⅱ相試験を省略の上で国際共同第Ⅲ相試験に組み入れることがあります。
このような国際共同治験がトレンドになっているので、最近は日米欧でほぼ同時に承認申請されることが多く、開発ラグも減ってきているように思います。
審査期間短縮のための施策

開発ラグ、審査ラグのいずれも解消している状況であることを紹介してきました。製薬企業としてもPMDAとしても、無駄な試験を実施せず効率的に開発が進むことを望んでいます。そのために、PMDAでは「対面助言」とよばれる治験相談を実施しています(FDAやEMAも同じような相談を実施しています)。
試験デザイン(対象患者、症例数、評価項目、統計解析など)をPMDAと合意した上で臨床試験を実施することで、承認申請後の試験のやり直しを防ぐことができます。例えば、第Ⅲ相試験の試験デザインを相談する「医薬品第Ⅱ相試験終了後相談」は950万円程度と高額ですが、臨床試験をやり直すことを考えると安いのか、毎月コンスタントに相談が申し込まれています。
対面助言の仕事

この対面助言、中の人の仕事としては以下のようなルーティンで進めています。
- 資料搬入(対面助言実施日から起算して5週前の月曜日)
- 照会事項の発出 → 1週間後に製薬企業から回答
- 機構意見の発出 → 1週間後に製薬企業から相談者見解
- 対面助言当日
まず、対面助言実施日から起算して5週前の月曜日に製薬企業から相談資料一式を受け取ります。これには、相談内容が具体的に記載されている「相談資料」、実施予定の臨床試験の詳細を説明した「治験実施計画書」、これまでの非臨床・臨床試験の結果をまとめた「治験薬概要書」など、何百〜何千ページにも及ぶ膨大な資料が添付されてきます。
その内容を確認し、説明不足や意味不明な点を「照会事項」としてまとめて製薬企業に聞きます。雑なダミーですが、以下のようなイメージです。

その後、相談資料の内容や照会事項に対する回答を踏まえ、相談内容に対するPMDAの意見を「機構意見」として製薬企業に送付します。その1週間後に、製薬企業として見解を「相談者見解」としてまとめてもらいます。PMDAの意見に合意するなら「機構の意見を了解した」、合意できないならその理由をつらつらと書いてもらう感じ。
そして、対面助言当日には、これまでの書面上のやりとりで合意できなかった部分を中心に、PMDAの会議室で直接会って内容を詰めていきます(コロナ禍ではWeb会議です)。
ひとつの相談ごとに、以上のようなルーティンを約5週間で回しています。このような相談が常にいくつも同時進行しているようなイメージです。
治験届調査
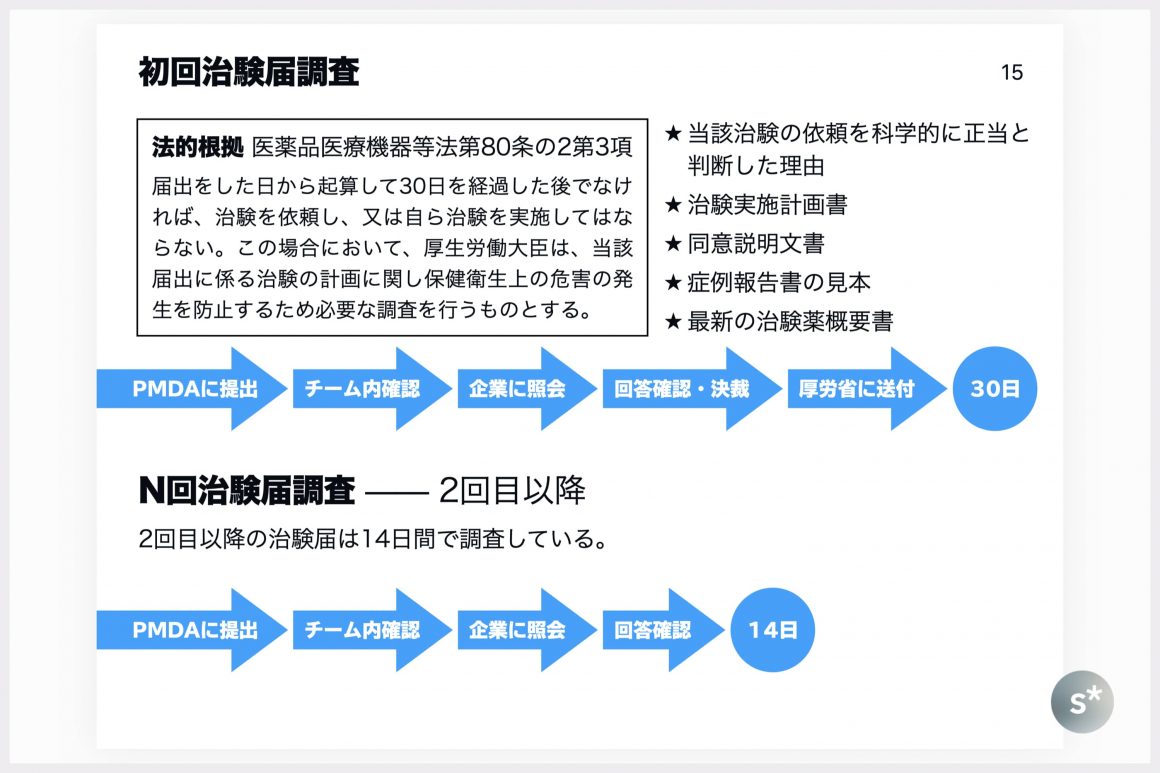
製薬企業が新たな治験を実施するときは、PMDAに対して届け出る必要があります。その届出の内容をPMDA内で調査し、安全上の懸念がある場合は計画を変更してもらったりすることもあります。
特に、日本で初めて治験を実施する治験薬の場合は「初回治験届調査」の対象となります。これは薬機法が根拠となる調査で、最終的には厚生労働大臣宛に調査結果を報告する必要があります。これは届出から30日以内に調査を終わらせる必要があるので、「30日調査」とよばれています。
その治験薬を用いた治験が2回目以降の場合は、2回目だったら2回治験届、3回目だったら3回治験届、というように、製薬企業は「○回治験届」をPMDAに提出します。この調査は必要に応じて14日間で行われ、「N回届調査」とよばれます。
承認審査

製薬企業が承認申請するときは、その資料を「CTD(Common Technical Document)」という形式に沿ってまとめなければなりません。これは日米欧で共通化されたフォーマットであり、第1部(モジュール1)〜第5部(モジュール5)から成っています。
提出されたCTDをもとに、審査は約1年かけて実施されます。具体的には「審査報告書」というものを作っていく作業であり、その審査報告書の作成の上でCTDに説明不足な点があれば照会事項を発出して追加説明を求めます。
また、前述の対面助言の実施は任意なので、全くの相談なしに承認申請することは制度上可能です。そのため、もし試験に不備があって説明されないと承認できないような重要な内容が欠如していた場合は、試験のやり直しを求める可能性もあります。ただ、少なくとも僕は聞いたことないので相当稀だと思います。
ちなみに、CTDを構成する臨床試験のまとまりのことを「臨床データパッケージ」とよびます。この臨床データパッケージに含まれている試験の結果をもとに、製薬企業がCTDにおいて薬剤の有効性や安全性を論じることになります。
臨床データパッケージは対面助言であらかじめ合意しておくことが多いです。例として、「タリージェ」という薬のCTDの一部がPMDAのサイトで公表されていたので持ってきました。ここまで多数の試験で構成されることは少ないですが、一例としてご参考にどうぞ。

学会参加と論文執筆
頻度は低いですが、自身が携わる可能性のある疾患領域の学会に参加することもあります。国内学会だけでなく海外で開催される国際学会にも参加できますが、コロナ禍でほぼオンライン開催になってしまいました。本当はホノルルに行く予定だったんですけどね…
また、学会参加よりもさらに頻度が低いですが、論文を書く人もいます。ほとんどの審査専門員は論文執筆とは縁のない生活をしていますが、学会から協力の要請があれば書くくらいです。
まとめ
以上をまとめると、PMDAの新薬審査部においては以下のような仕事をしており、日本の薬事規制を担っています。
- 審査
- 対面助言(治験相談)
- 治験届調査(初回、N回)
- 学会参加、論文執筆
最後に、気になる「残業」について少しだけ書いておきますね。PMDAはブラックだと言われることが多いように思いますが、これは本当に人によります。各審査専門員にほぼ均等に案件が割り振られるので、効率的に案件を捌けば残業は少ないです。僕は月に10〜20時間くらいでした。
と、PMDAの新薬審査部における仕事内容をご紹介しました。薬学生の方から質問があれば答えるので、Twitterでリプライでも飛ばしてください〜







